
A Closer Look: The History and Authority of the FDA
In April of 2023, a U.S. District Judge ruled to suspend FDA approval of mifepristone, a synthetic steroid approved by the agency in 2000 for use in drug-induced abortions and the treatment of Cushing's syndrome. This ruling, discounting the FDA’s role in consumer safety and the scientific data they used when evaluating the product, caused concerns about the agency’s independent regulatory authority — and the future of science-based evaluation of products in the U.S. So, we thought it would be useful to take a look at the history of the FDA and why they were granted this authority in the first place.
Precursors and the birth of the FDA
Some food regulation came with the birth of the U.S., and over the years, a patchwork of regulatory initiatives and laws followed:
- 1813: The Vaccine Act of 1813 was a federal program developed to address one of three issues limiting success with smallpox vaccinations in the U.S. at the time: public opposition, expense of vaccination, and lack of reliable supply. Supply was an issue for two main reasons. First, people clamored for vaccination only during periodic smallpox epidemics. As such, states would not take on the expense associated with maintaining a reliable supply of cowpox. Second, pharmaceutical companies were yet to evolve. For these reasons, the Federal government stepped in to ensure consistent availability of smallpox vaccine. Dr. James Smith was named the first National Vaccine Agent. The law was repealed in 1822 after an incident known as the “Tarboro Tragedy.” Sixty people fell ill and 10 died from a smallpox epidemic introduced in the North Carolina community when Dr. Smith accidentally sent smallpox instead of cowpox to the local “auxiliary vaccine agent,” named Dr. Ward. Failing to recognize the error, Dr. Ward inoculated townspeople seeking vaccination, thereby causing the outbreak. Politicians eager to end the program quickly used the incident as a reason to do so.
- 1820: The U.S. Pharmacopeia was developed by an independent group of 11 physicians determined to standardize medications. They wanted to protect their patients from unsound “miracle cures” and medication errors resulting from the variety of preparation methods used by local apothecaries. Using the latest science, the group met in Washington, D.C. and created the first edition of the U.S. Pharmacopeia. They sought to remain separate from the government and hoped to build trust in traditional medicine, which at the time was often dismissed in favor of advice from midwives, folk healers, and Native American shamans
- 1848: During the early 1800s, the U.S. was importing many medications, which were often of sufficiently poor quality that they could not be sold in Europe. As this issue came to attention, Congress passed the Drug Importation Act of 1848 to prohibit impure and weak drugs from entering the country. The Act relied on the standards set by the U.S. Pharmacopeia, setting the stage for future regulation of drugs.
Although these initiatives and others helped to increase consumer safety, no wide-ranging regulatory authority existed until the beginning of the 20th century when the 1906 Pure Food and Drugs Act and the Meat Inspection Act were established. These laws, which outlawed the interstate sale of medications, foods, and beverages that were not accurately branded, resulted from revelations of unsanitary practices in meat-packing plants, use of poisons as additives in food, and false claims used in promoting “cure-all” products. The responsibilities outlined in the Pure Food and Drugs Act were originally under the purview of the U.S. Department of Agriculture’s (USDA) Bureau of Chemistry. However, in 1927, the regulation-related responsibilities of the Bureau of Chemistry were moved to a newly created USDA agency, called the Food, Drug, and Insecticide Administration. A few years later, in 1930, the name was adjusted to the Food and Drug Administration (FDA).
The "American Chamber of Horrors"
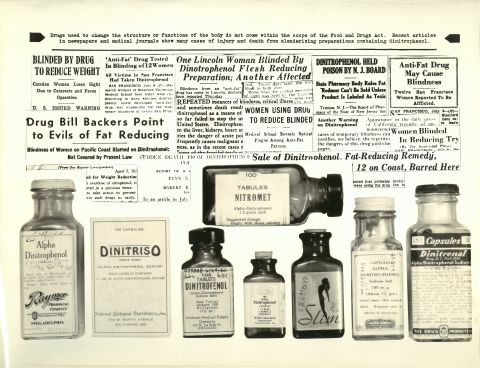
The 1906 Pure Food and Drugs Act laid the foundation for consumer safety regulation by necessitating truthful labeling and requiring products to meet purity and strength standards. Unfortunately, the law also had many loopholes, including strict limits on which products could be regulated, high burdens of proof for untruthful labeling, and limited authority to remove harmful products from market.
In 1933, two FDA employees, Chief Education Officer, Ruth deForest Lamb, and Chief Inspector, George Larrick, designed a traveling exhibition to make the public aware of the regulatory shortcomings of the law and the associated dangers. The exhibit displayed ineffective and toxic products that the FDA could not prohibit. Many of the examples in the show were so disturbing that one journalist described the exhibit as the “American Chamber of Horrors.”
Although the show had a major impact on the American public and legislators, it wasn’t until several years later that some much-needed adjustments were implemented. Unfortunately, during those intervening years, more people were harmed or killed by dangerous products. One famous, or perhaps infamous, example is the so-called “Sulfanilamide Disaster,” which occurred in 1937. More than 100 unassuming patients died after taking a medicine containing diethylene glycol, commonly used as antifreeze. This ingredient was added to sulfanilamide to create a liquid version of a popular medication used to treat bacterial infections. However, at the time, safety studies were not required, so while the company had tested the new formula for flavor, appearance and fragrance, they had not tested its toxicity. In response to this tragedy, a new law, called the 1938 Federal Food, Drug, and Cosmetic (FDC) Act, was passed, finally expanding the FDA’s regulatory authority to protect the American consumer.
Milestones, successes and challenges
Over the decades, the FDA’s responsibilities expanded in reaction to developments in medicine and technology. Expanded protections related to drugs, infant formula, blood donations, food packaging, vaccines, medical testing, tobacco products, bioterrorism preparedness, and more. One major success in drug regulation occurred in 1962 when an FDA medical officer, Dr. Frances Kelsey, blocked the use of thalidomide in the U.S. Thalidomide had been prescribed for sleep and morning sickness in Europe, where it was discovered to have caused birth defects in babies of women who took it.
Even with testing and oversight, sometimes the FDA must establish new rules and regulations in reaction to a previously unanticipated threat, such as in the 1980s when it required tamper-proof packaging for over-the-counter medications. This action was in response to a crime in which several people were killed after taking Tylenol that a local resident had laced with cyanide.
Even after a drug or product is approved, the work of FDA regulators does not end. They continue to review new data and situations that arise. On occasion, this means products are removed from the market or updated warning labels or recommendations associated with their use are required. Examples include the removal of Vioxx, a pain medicine, when data revealed that it could increase the risk of cardiovascular disease, and Belviq, a weight-loss medication, which was linked to an elevated risk of cancer. It is important to note that these medications were removed after rigorous scientific evaluation indicated the risks outweighed the benefits, in contrast to the recent ruling with mifepristone, which ignored the FDA data evaluation process that the public and industry rely upon. In an open letter responding to the mifepristone ruling, many pharmaceutical scientists and leaders noted, “As an industry we count on the FDA’s autonomy and authority to bring new medicines to patients under a reliable regulatory process for drug evaluation and approval.”
In sum
Throughout history, the FDA has implemented laws and regulations to protect against dangerous or ineffective products, respond to medical and technological advancements, and address public health emergencies. One thing that has remained constant throughout the years is the FDA’s steadfast commitment to science and research, and the reliance on quality data to chart the course. Now, questions loom for the future. What will happen if the agency’s regulatory authority is taken away? Will Americans again find themselves with nothing standing between them and dangerous products?
Related resources
For a look behind the scenes at the FDA, watch Marion Gruber: Preparedness is Prevention, a 30-minute film highlighting the years Marion spent working at the FDA to ensure vaccine safety and address infectious disease outbreaks.
For more details on the evolution of the FDA and what incidents or legal cases compelled its expanded role in consumer protection, see their webpage, “Milestones in U.S. Food and Drug Law.”
Also check out these other resources:
- Missed Opportunities: The Vaccine Act of 1813 (research paper)
- “From guesswork to standards: The history of medicine quality” (article)
- 1848 Drug Importation Act (article)
- 80 Years of the Federal Food, Drug, and Cosmetic Act (online exhibit)
- "The American Chamber of Horrors" (video)